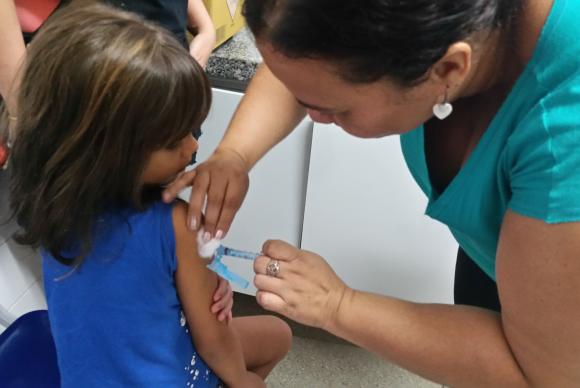
Pesquisadores do Instituto Oswaldo Cruz (IOC/Fiocruz) finalizaram o sequenciamento completo do genoma do vírus responsável pelo atual surto de febre amarela no país. A partir dessa análise, eles encontraram variações inéditas em algumas de suas sequências genéticas. Não há registro anterior dessas mutações na literatura científica mundial, de acordo com a instituição.
A equipe de cientistas informa, no entanto, que a vacina usada atualmente protege contra diferentes genótipos do vírus, incluindo o sul americano e o africano, e que as alterações detectadas no estudo não tiram a eficácia de quem tomou uma dose.
“A vacina vai proteger certamente. Um exemplo disso é que em qualquer lugar do mundo que você tem variantes da febre amarela, a vacina protege com a mesma eficácia. A princípio não muda nada”, disse uma das pesquisadoras, Myrna Bonaldo.
Esse é o maior surto de febre amarela das últimas décadas. O último boletim do Ministério da Saúde confirmou 756 casos no país, com 259 mortes devido à infecção. Os casos continuam silvestres, com infecções em regiões de mata e/ou rurais. A doença é transmitida pelos mosquitos Sabethes e Haemagogus.
O sequenciamento
Desde o aumento de casos no Brasil, a Fiocruz fez os primeiros sequenciamentos do vírus. Foram utilizadas duas amostras de macacos bugios do Espírito Santo, mortos em fevereiro de 2017. Os resultados foram publicados na revista científica “Memórias do Instituto Oswaldo Cruz”.
“Os macacos bugios são especialmente importantes nas investigações sobre a febre amarela por serem considerados ‘sentinelas’: como são muito vulneráveis ao vírus, estão entre os primeiros a morrer quando afetados pela doença. Além disso, estes animais amplificam eficientemente o vírus em seu organismo”, descreve Ricardo Lourenço, que é veterinário e entomologista.
Um resultado inicial apontou que esse vírus da febre amarela pertence ao subtipo genético conhecido como linhagem Sul Americana 1E, que atua no Brasil desde 2008. No entanto, com o final da análise completa, os cientistas conseguiram detectar as variações genéticas, que estão associadas a proteínas envolvidas na replicação viral.
De acordo com os pesquisadores, os impactos da descoberta para a saúde pública ainda precisam ser investigados e apontam a necessidade de que mais amostras sejam sequenciadas, relativas a outros lugares do Brasil e com coletas em humanos, macacos e mosquitos. Novos resultados deverão ser apresentados nas próximas semanas.

Primatas são as principais vítimas da febre amarela (Foto: Arquivo TG)
Os resultados da pesquisa foram encaminhados pela presidência da Fiocruz ao Departamento de Vigilância das Doenças Transmissíveis, do Ministério da Saúde. De acordo com Bonaldo, os resultados também foram encaminhados para a comunidade internacional, incluindo Itália, Estados Unidos e Inglaterra.
A instituição informa, adicionalmente, que outros dados ainda não publicados apontam os mesmos resultados para a análise de mosquitos coletados no Espírito Santo e para um macaco morto no Rio de Janeiro.
A fundação diz, ainda, que o estudo “partiu de uma constatação que vem ganhando cada vez mais espaço”. Segundo eles, “a atual situação de febre amarela tem lacunas de entendimento sobre sua dinâmica de dispersão.”
Hipóteses de mutação
Segundo Bonaldo, uma possibilidade para a mutação ter ocorrido é a capacidade do vírus se modificar geneticamente com frequência (não tanto como o da gripe), e também devido à baixa cobertura vacinal antes do surto na região do Rio de Janeiro e Espírito Santo.
A pesquisadora também detalhou como serão os próximos passos do estudo e destacou que ele serve para conhecer a capacidade circulação do vírus no país.
“O estudo dá ferramentas preciosas para fazer uma melhor vigilância sanitária e prever piores casos, além de saber que regiões do Brasil podem ser priorizadas na hora de uma vacinação”, disse.
A partir de agora, a equipe irá estudar o vírus em laboratório e comparar o tipos anteriores ao surto atual. Também tentarão estabelecer se esse caso pode ser mais agressivo ou não.
Bonaldo disse que os pesquisadores estão com mosquitos selvagens e urbanos, uma busca por entender se eles possuem o mesmo potencial de infecção.
“Vamos poder conhecer um pouco mais da biologia do vírus”, afirmou.
Fonte: TH